Since 2008, the dramatic expansion of genomic studies of ACC has led to a deeper understanding of the disease’s underlying biology.
 Here we attempt to summarize the molecular, preclinical and clinical evidence for the importance of various biomarkers implicated and/or targetable in ACC. For each molecular target, we organize the evidence into the following categories:
Here we attempt to summarize the molecular, preclinical and clinical evidence for the importance of various biomarkers implicated and/or targetable in ACC. For each molecular target, we organize the evidence into the following categories:
Molecular evidence
– DNA
– RNA/protein
Preclinical evidence
Clinical evidence
In descending order of perceived significance, these molecular targets are:
The MYB proto-oncogene is a transcription factor that regulates stem and progenitor cells in the bone marrow, colonic crypts, and brain. MYB and its family members (a-MYB/MYBL1 and b-MYB/MYBL2) contain an N-terminal DNA-binding domain, central transcriptional activation domain, and C-terminal domain that direct substrate specificity and protein-protein interactions. c-MYB and a-MYB, but not b-MYB, share highly homologous DNA binding and transactivation domains.
The MYB gene is frequently upregulated via chromosomal rearrangement or amplification in a variety of cancers (leukemia, lymphoma, breast, colon, angiocentric glioma, and adenoid cystic carcinoma). C-terminal truncation of MYB is thought to unmask its oncogenic potential in vitro (Zhou and Ness, 2011). Truncations involving MYB (Bandopadhayay et al., 2016) and MYBL1 (Ramkissoon et al., 2013) are found in pediatric gliomas.
Molecular evidence
DNA
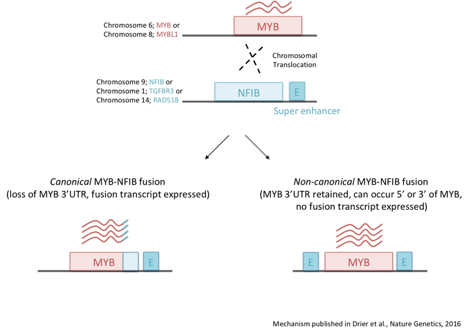
- The t(6;9) translocation, resulting in a MYB-NFIB gene rearrangement, occurs in 50-60% of ACC tumors, making it the most frequent, recurrent alteration in ACC. The t(6;9) translocation site is promiscuous and, in some cases, may lead to the generation of an MYB-NFIB fusion gene (Persson et al., 2009, Mitani et al, 2010, Brill et al., 2011, Mitani et al., 2011, West et al., 2011 and Persson et al., 2012).
- MYB-NFIB rearrangements have been detected in ACCs originating from various body sites (salivary, lacrimal gland, breast, etc.) and are specific to ACC, except for sporadic dermal cylindromas, which share some histological features of ACC tumors (Fehr et al., 2011).
- Chromosomal rearrangements involving MYB and other genes (TGFBR3, RAD51B, EWSR1) occur in approximately 15% of ACC tumors (West et al., 2011, Drier et al., 2016 & Lei et al, 2023).
- Genomic translocations involving MYBL1 (MYB’s homolog) and NFIB (or other genes like YTHDF3, RAD51B) are observed in 10-30% of ACCs and occur mutually exclusive with alterations in MYB. Likewise, ACCs with MYBL1 rearrangements display high levels of MYBL1 and low levels of MYB protein expression (Mitani et al., 2016 & Brayer et al., 2016). MYBL1 rearrangements have also been reported in ACC of the breast (Kim et al., 2018), tracheobronchial (Pei et al., 2019), and skin (Kyrpychova et al., 2018). Some evidence suggests that rearrangements involving MYBL1 may frequently occur in the mandibular region (Endo et al., 2019).
- Approximately 30% of MYB rearrangements disrupt C-terminal sequences involved in negative miRNA-mediated regulation of MYB and protein-protein interaction specificity (Persson et al., 2009, Mitani et al, 2010 & Drier et al., 2016).
- A unifying feature of all MYB rearrangements in ACC is the juxtaposition of super-enhancers near the MYB locus. Displaced enhancers are bound by the MYB protein and interact with MYB’s promoter to drive a positive feedback loop that sustains high MYB expression (Drier et al., 2016).
- High-level MYB gene amplification has been detected in breast ACC (Kim et al., 2018).
- A large, multi-institutional analysis of over 400 ACC cases revealed MYB/MYBL1 alterations in 78% of cases (62% with MYB; 16% with MYBL1) and overexpression of MYB/MYBL1 in 93% of cases. Loss of the 3’ end of MYB was found to be enriched in grade 3 tumors and a predictive biomarker of poor overall survival (Persson et al., 2022).
- MYB/MYBL1- and peri-MYB/MYBL1 (defined as centromeric and telomeric areas of 10 Mb each)-associated rearrangements were observed in 93% (149/160) of salivary adenoid cystic carcinoma cases. Tumors with peri-MYB-associated rearrangements overexpressed MYB (via qRT-PCR or IHC), similar to tumors with MYB-associated rearrangements (Ueda et al., 2023).
- Gene fusions between MYB and TULP4, ACTN4, EWSR1, and ACTB have also been detected at low frequency in ACC tumors (Skalova et al., 2024).
 RNA/protein
RNA/protein
- Overexpression of MYB/MYBL1 is observed in 80-90% of ACCs (Gao et al., 2014, Brayer et al., 2016 & Frerich et al., 2019. Lack of reliable MYB and MYBL1 antibodies engineered against epitopes retained in ACC tumors for IHC and other assays has hindered a definitive analysis.
- MYB or MYBL1 fusion transcripts are detected in many MYB or MYBL1-rearranged tumors. Evidence for alternative splicing of MYB is suggested by the presence of multiple fusion transcripts or both full-length and truncated forms of MYB in a single ACC tumor (Mitani et al., 2010 & Brayer et al., 2016). N-terminal truncated versions of MYB protein produced via an alternative MYB promoter have also been reported in ACC tumors (Frerich et al., 2019).
- ACC tumors harboring either MYB or MYBL1 alterations share a common gene signature, suggesting interchangeable oncogenic functions of the two transcription factors. Combined MYB and MYBL1 expression is correlated with poor clinical outcomes in ACC patients (Mitani et al., 2010, Mitani et al., 2011, Gao et al., 2014 & Brayer et al., 2016).
- A list of putative MYB target genes has been nominated based on MYB chromatin binding profiles and differential gene expression in ACC patient-derived xenograft (PDX) tumors versus normal salivary tissue. This list includes the following genes: MYC, TP63, BCL2, AURKA, CCND1, MET, FGFR2, IGF1R, MALAT1, CASC4, NENF, CDK6, GMNN, NOTCH1 and NOTCH pathway genes (JAG1, JAG2 and SPEN) (Drier et al., 2016).
Preclinical Evidence
Preclinical evidence that MYB initiates ACC tumorigenesis:
- Three MYB-NFIB genetically engineered mouse models (GEMMs) have been developed.
- Mice engineered to express the MYB-NFIB transgene (MYB exons 1-14 fused to NFIB exon 12) under control of Cre-activated MMTV promoter show upregulated transgene expression (by RT-PCR) in salivary and breast tissues and develop B-cell neoplasms, but not salivary tumors, potentially stemming from lack of MYB-NFIB overexpression within the salivary gland. Crossing of MYB-NFIB/MMTV-CRE and Ink4a+/-/Arf+/- mice (modeling p16 inactivation that is observed in some ACCs) led to the formation of a large breast tumor with ACC-like histology that expresses high levels of MYB protein (Jiang et al., 2019).
- Mice engineered to express the MYB-NFIB transgene (MYB exons 1-14 fused to NFIB exon 8) under control of the Cre-activated MMTV promoter did not show high levels of MYB protein expression in the salivary gland (even upon direct injection of adenovirus-Cre directly) and failed to form tumors of any kind. Crossing of MYB-NFIB/MMTV-CRE mice with the p53fl/fl mouse model led to the formation of hyperplastic glands that developed into poorly differentiated breast adenocarcinomas in mice with heterozygous loss of p53 and lymphoma in mice with homozygous loss of p53. Breast tumors forming in the MYB-NFIB/MMTV-CRE/p53fl/+ mice showed elevated levels of MYB and keratin protein but lacked classical ACC histology (Mikse et al., 2016).
- Tet-inducible, MMTV-driven full-length, wild-type MYB or MYB-NFIB transgenes can both drive ACC tumor formation in mice with slightly different penetrance and latency characteristics. ACC tumors that form in MYB-driven GEMMs depend on MYB expression for sustained growth. Crossing of Tet-inducible, MMTV-driven MYB overexpressing mice with a Tet-inducible, MMTV-driven NOTCH-active GEMM accelerates tumor formation in vivo (work presented by Dr. Carlos Caulin at the 2019 Advances in ACC Translational Research Conference; also AACR 2020 poster).
Preclinical evidence that MYB is a therapeutic target in ACC:
- MYB-NFIB suppression using genomic or chemical reagents inhibits the proliferation of patient-derived ACC cells in vitro (Andersson et al., 2017, Jiang et al., 2019 & Yusenko et al., 2020).
- Treatment of ACC PDX models with RGT-61159, a novel mRNA degrader of MYB, significantly reduced tumor growth and MYB protein levels (Xi et al., 2024).
- Naturally occurring compounds (for example Naphthol, Celastrol, Teniposide, Etoposide, and Monensin) and all-trans retinoic acid (ATRA) have been identified in vitro and in vivo to inhibit MYB and exert anti-proliferative effects in short-term culture ACC cells and PDX models (Uttarkar et al., 2015, Uttarkar et al., 2016, Yusenko et al., 2018, Yusenko et al., 2020 and Mandelbaum et al., 2018). A small, phase 2 clinical trial testing ATRA in 18 recurrent/metastatic ACC patients showed stable disease as the best response in 61% of evaluable patients (Hanna et al., 2021).
- Treatment of low-grade ACC PDXs with the BET bromodomain inhibitor JQ1 inhibits tumor growth and MYB and MYB target gene expression (Drier et al., 2016).
clinical Evidence
- A phase 1 trial (NCT06118086) testing REM-422, a small-molecule mRNA degrader of MYB, in recurrent/metastatic ACC patients is recruiting.
- A phase 1 trial testing a DNA MYB vaccine in combination with a PD-1 inhibitor in recurrent/metastatic ACC patients completed recruitment; results are pending.
In mammalian cells, the NOTCH pathway consists of four NOTCH receptors (NOTCH1-4) that harbor a short extracellular region of EGF-like repeats, a single transmembrane-pass region and a small intracellular region. Binding of NOTCH ligands, Delta-like 1 (DLL1), DLL3, DLL4, JAGGED 1 (JAG1) or JAG2, to their receptor(s) triggers a series of proteolytic events by the ADAM-family metalloprotease and presenilin-gamma-secretase that release the NOTCH intracellular domain (NICD) from the plasma membrane and allows nuclear translocation. Upon entering the nucleus, NICD converts SPEN-bound CSL/RBPJ transcriptional repressor complexes into transcriptional activating complexes by recruiting the Mastermind (MAML1) co-activator protein to drive gene expression programs (classic targets include: HES, HEY, CCND1, MYC) linked with stem cell function and development in a variety of tissues, including brain, cardiovascular, pancreas, intestine and bone. The pathway is then turned off following phosphorylation of NICD within its PEST domain, which targets it for ubiquitin proteasomal degradation by FBXW7.
Aberrant NOTCH pathway activation has been implicated in a variety of cancers (including breast, colorectal, pancreatic, lung, etc). Paradoxically, the NOTCH pathway has also been shown to exert tumor suppressive functions in certain cell contexts (breast, prostate, glioblastoma, etc.). A number of complexities have been proposed to contribute to NOTCH’s pro- or anti-tumorigenic functions, including tumor microenvironment and cross-talk between other signaling pathways. In addition to tumor initiation, NOTCH pathway deregulation has also been linked to tumor progression and drug resistance.
Molecular evidence
DNA
- Approximately 9% of primary and up to 27% of recurrent/metastatic ACC patients harbor a mutation in the NOTCH1 gene (Ho et al., 2013, Stephens et al., 2013, Ross et al., 2014, Mitani et al., 2016, Ferrarotto et al., 2016, Rettig et al., 2016 and Ho et al., 2019).
- Gain-of-function (activating) mutations in NOTCH1 occur in approximately 18% of patients with recurrent/metastatic disease (Ho et al., 2019). Activating NOTCH1 mutations occur in hotspot regions (the negative regulatory region and Pro-Glu-Ser-Thr-rich (PEST) domain) detected in other cancer types and are predicted to confer sensitivity to the gamma-secretase class of NOTCH inhibitors (Ferrarotto et al., 2016). Activating rearrangements in the NOTCH1 allele have also been detected in ACC (Sajed et al., 2017).
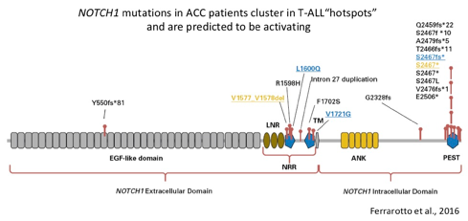
- Mutations in the NOTCH2, NOTCH3 and NOTCH4 genes, as well as other members of the NOTCH pathway (SPEN and FBXW7) have also been observed in ACC (Ho et al., 2013, Stephens et al., 2013, Ross et al., 2014, Mitani et al., 2016, Rettig et al., 2016 and Ferrarotto et al., 2016) and trend towards enrichment in recurrent/metastatic disease. Approximately 24% of recurrent/metastatic cases have a mutation in NOTCH1-4 and almost 40% have a mutation in a NOTCH pathway gene (NOTCH1-4, SPEN or FBXW7; Ho et al., 2019).
- NOTCH1 mutations correlate with solid histology and define an aggressive disease subset associated with higher risk of relapse (12.5 months in NOTCH1-mutant compared to 33.6 months in NOTCH1-wt) and shorter overall survival (29.8 months in NOTCH1-mutant compared to 122 months in NOTCH1-wt) (Ferrarotto et al., 2016 and Zhang et al., 2020). Poorer survival of patients with activating NOTCH1 mutations has been confirmed in a larger patient cohort (>1K patients; median OS 31.1 months in NOTCH1-mutant vs. 73.8 months in NOTCH1-wt; Ho et al., 2019).
- NOTCH1 mutations co-occur with mutations in other NOTCH pathway genes (Ferrarotto et al., 2016) and with chromatin-modifying genes KDM6A, ARID1A and CREBBP (Ho et al., 2019). Simultaneous introduction of two NOTCH1 mutations in vitro led to hyperactivation of a NOTCH1 reporter assay (Ferrarotto et al., 2016).
- NOTCH1 mutations were found to be mutually exclusive with TERT promoter mutations (Ho et al., 2019).
- NOTCH mutations are typically truncal and preserved in metastatic lesions (Liu et al., 2017) and Ho et al., 2019). Clonal enrichment for NOTCH1 mutations has been observed during progression of adenoid cystic carcinoma of the breast to triple-negative breast cancer (Fusco et al., 2016).
- In a UK cohort of 120 ACC patients (95% recurrent/metastatic), NOTCH activation was observed in 11% (13/120) and NOTCH mutant patients had a more aggressive disease course compared to patients with wild-type NOTCH (median recurrence-free survival 1.1 vs. 3.4 years, median overall survival from diagnosis 4 vs. 16.3 years and median overall survival from time of disease recurrence/metastasis 1.9 vs. 9.6 years; Feeney et al., 2022).
RNA/protein
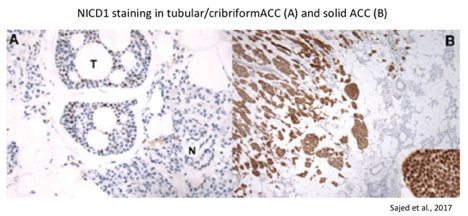
- Virtually all ACCs stain positive for the intracellular domain of NOTCH1 (NICD; activated form) and fall into two staining patterns: subset- or diffuse-positive. Diffuse-positive NICD staining correlates with the presence of NOTCH1 gain-of-function mutations and is restricted to ACC tumors with solid histology. Diffuse NICD staining is associated with worse outcome (median survival of 56.6 months in diffuse-positive vs. 140 months in subset-positive; Sajed et al., 2017 and Bell et al., 2014). Most solid ACCs show NICD overexpression, whereas very few cribriform-tubular tumors do (Zhang et al., 2020).
- MYB drives a NOTCH-associated transcriptional program in the luminal/epithelial cell compartment of low-grade ACC tumors (Drier et al., 2016).
- Due to cell-type specific differences in NOTCH signaling, the downstream targets of NOTCH1 in ACC are still largely unknown. For example, HES4 expression does not correlate with NOTCH activation in ACC PDXs (Stoeck et al., 2014).
- RBPJ transcription factor sites are enriched for H3K27 acetylation (histone mark of enhancer regions) in grade 3 ACC PDXs (Drier et al., 2016). It is unclear whether NOTCH-independent RBPJ effects contribute to ACC progression as has been observed in brain tumor-initiating cells in glioblastoma (Xie et al., 2016).
- Diffuse staining of the NOTCH target gene FABP7 predicts poor ACC patient prognosis (Phuchareon et al., 2014 and Phuchareon et al., 2018).
Preclinical Evidence
- Activating NOTCH1 mutations (NRR region) confer sensitivity to gamma-secretase inhibitors (GSIs) in ACC PDX models (Stoeck et al., 2014).
- GSI treatment (DAPT) reduced NOTCH-positive ACC cell proliferation and sphere formation in vitro and tumor growth in vivo (Panaccione et al., 2016).
- Brontictuzumab, a NOTCH1 neutralizing antibody, significantly inhibited tumor growth in a NOTCH1-mutant ACC PDX model (Ferrarotto et al., 2016).
- NOTCH inhibition via treatment with AL101 (GSI) inhibited the growth of patient-derived, NOTCH-mutant ACC cells grown as organoids and PDXs. AL101 treatment led to a reduction in NICD1, MYC and Ki67 levels in NOTCH-mutant PDX tumors, as well as reduced the expression of 21 NOTCH-related genes (see complete gene list in Ferrarotto et al., 2022).
Clinical Evidence
- One ACC patient with a tumor harboring 2 activating NOTCH1 mutations achieved a partial response when treated with the NOTCH1 antibody, Brontictuzumab. Additional NOTCH pathway mutations were detected in a newly developed metastasis following patient progression, suggesting a potential mechanism of acquired resistance (Ferrarotto et al., 2016).
- A phase 1 trial testing the gamma-secretase inhibitor LY3039478 in 22 recurrent/metastatic ACC patients (unselected with unknown NOTCH mutational status) led to 1 (4.5%) partial response. The disease control rate was 16/22 (73%) patients, of which 4 (18%) patients exhibited stable disease lasting >= 6 months. The median PFS was 5.3 months (Even et al., ASCO abstract 2017). (Even et al., ASCO abstract 2017).
- One partial response was observed in a NOTCH1-mutant ACC patient treated with the gamma-secretase inhibitor AL101 (formerly BMS-906024) (El-Khoueiry et al., ASCO abstract 2018).
- Preliminary efficacy results from the ACCURACY phase II clinical trial testing AL101 in 77 recurrent/metastatic ACC patients with activating NOTCH mutations showed a partial response rate of 12% and stable disease in 57% of patients for >2 months (Ferrarotto et al., ESMO 2019 and Ferrarotto et al., ASCO 2022).
- A phase 1 trial testing the CSL-NICD gene transcription complex inhibitor CB-103 in 19 ACC patients (unselected for NOTCH pathway status) led to stable disease as the best response. The median PFS observed in ACC patients was 21.7 weeks and the disease control rate was 79% at week 8 and 58% at week 20. Three NOTCH-active ACC pts had radiologically confirmed SD at >6 months (Miranda et al., ASCO abstract 2021).
- One tracheal NOTCH1-mutant ACC patient experienced a significant reduction in tumor burden (meeting partial response RECIST criteria) and cancer-related symptoms while taking the GSI Nirogacestat. The patient displayed 6.5 months of progression-free survival and remained on drug for 400 days, with an apparent clinical benefit over that time. Tumor profiling of a biopsy taken from a metastatic lung lesion revealed two NOTCH1 mutations, NOTCH1 gene amplification and mutations in two NOTCH inhibitor genes, SPEN and TSC1 (Kieran et al., 2021.)
- A phase I window-of-opportunity study(NCT04973683) testing NOTCHi AL101 in NOTCH-active ACC patients prior to surgery of their primary tumor is currently recruiting.
- A phase I/II study (NCT05774899) testing CB-103 + Lenvatinib (multi-TKI) or Abemaciclib (CDK4/6 inhibitor) is currently enrolling ACC patients with activating NOTCH mutations.
Receptor tyrosine kinases are a subset of protein kinases involved in regulating cellular responses to growth stimuli, including cell growth, cell proliferation, stromal growth, angiogenesis and tissue invasion. Transmembrane tyrosine kinases consist of an extracellular domain that upon ligand binging triggers an activating conformational shift and release of auto-inhibitory mechanisms. Dimerization and trans-phosphorylation events are often involved in protein kinase activation and signal amplification. Individual tyrosine kinase signaling cascades often converge on downstream master regulators of growth, including the RAS/MAPK, PI3K/AKT, and PKC pathways.
Tyrosine kinase genes are frequently altered or upregulated in cancer, leading to constitutive or hyperactivation of their corresponding signaling pathways. Multi-kinase inhibitors are a class of drugs that target a number of different tyrosine kinase proteins with varying affinity (Du et al., 2018).
Molecular Evidence
DNA
- Several tyrosine kinase genes (FGFR-1, -2, -3, -4, VEGFR-1, -2, PDGFR-A, -B) have been found to be mutated or amplified in a small percentage (1-7%) of ACC tumors (Ho et al., 2013, Ross et al., 2014, Stephens et al., 2013 and Ho et al., 2019). Mutations in c-KIT are rare in ACC tumors (Moskaluk et al., 2010).
- Computational analysis of biological pathways disrupted by chromosomal rearrangement or somatic mutation suggests that altered FGF signaling is observed in a high percentage of ACCs (15/25 tumors; Rettig et al., 2016).
- Two (of 3) ACC patients with 4q12 amplification (containing PDGFR/KDR/KIT genes) achieved stable disease for >6 months while treated with axitinib (Ho et al., 2016).
- One ACC patient with a FGFR-2 mutation experienced a significant tumor response after treatment with Futibatinib, an irreversible, selective inhibitor of FGFR1-4 (Rodriguez et al., 2024).
RNA/protein
- Increased levels of FGF1, FGF2, FGFR1 and phosphorylated FGFR1 are frequently observed in ACC primary tumors (Myoken et al., 1996 and Gao et al., 2014) and PDX tissues (personal communication).
- All ACC tumor biopsies collected and analyzed as part of a phase II clinical trial for sorafenib showed expression of PDGFRb in the stroma and VEGFR2 in endothelial cells (Locati et al., 2016).
- The majority of ACC tumors express VEGF (88%) and KIT (94%) (Lee et al., 2012).
- Epithelial cells in ACC tumors are positive for KIT protein expression. In line with this, ACC tumors with solid histology show an enrichment for KIT-positive cells (Holst et al., 1999, Mino et al., 2003, Freier et al., 2005 and Salehinejad et al., 2014).
- In a 2022 literature review examining HER2/neu expression across different types of salivary gland cancers, 17% of ACCs were reported to express some level of HER2/neu expression (Javaheripor et al., 2022). High heterogeneity of HER2/neu overexpression rates across ACC publications may be partially linked to the finding that ERBB2 and ERBB3 expression is particularly strong in the invasive areas of tubular and cribriform ACC tumors (Shintani et al., 1995).
Preclinical Evidence
- Multi-tyrosine kinase inhibitors targeting FGFR, VEGFR, and PDGFR exert anti-tumorigenic activity in ACC PDXs (ACCRF-generated data and Younes et al., 2006).
- ACC PDX tumor cells (implanted in mice and zebrafish) are sensitive to regorafenib (Chen et al., 2017).
- FGFR1 signaling is increased in lacrimal ACC cells derived from patients treated with chemotherapy. A combination of cisplatin and the FGFR inhibitor AZD4547 synergistically reduces lacrimal ACC cell viability in vitro (Doddapeneni et al., 2019).
Clinical Evidence
More than a dozen clinical trials of multi-tyrosine kinase inhibitors in ACC have reported results. They are listed at the top of our Completed Studies page.
Recent studies of Lenvatinib and Apatinib (also named Rivoceranib) have demonstrated the highest rates of objective responses in ACC patients. Targeted drugs such as Axitinib and Sorafenib also appear to stabilize ACC tumors in a large proportion of ACC patients. These drugs are not cures but may keep the disease from progressing for 6 months or longer – an important benefit for patients with progressive disease. ASCO clinical guidelines suggest the use of multitargeted tyrosine kinase inhibitors such as Lenvatinib, Sorafenib, Axitinib, or Pazopanib if clinical trials are not available. NCCN clinical guidelines point to Lenvatinib as a reasonable option.
MDM2 (Mouse Double Minute 2 Homolog) is an E3 ubiquitin ligase that negatively regulates the p53 tumor suppressor gene by targeting it for proteolytic degradation. In response to DNA damage or other cellular stressors, MDM2 and p53 undergo phosphorylation that prevents their association and leads to stabilization and subsequent activation of p53 anti-tumorigenic responses (cell cycle arrest, DNA repair, senescence or apoptosis). p53-driven responses are limited by an autoregulatory negative feedback loop, whereby high levels of p53 trigger MDM2 expression.
MDM2 amplification or missense mutations that disrupt MDM2 binding to p53 have been reported in cancers with wild-type p53 and lead to p53 inactivation. MDM2 inhibitors have been proposed as a therapeutic strategy in cancers with wild-type p53.
Molecular Evidence
DNA/protein
- The majority of ACC tumors lack mutations in the TP53 tumor suppressor gene (Ho et al., 2013, and Martelotto et al., 2015) and express high levels of MDM2 (de Araujo et al., 2000, Jia et al., 2004, and de Lima et al., 2009 ). MDM2 amplification and mutation have also been reported in a subset of ACC tumors (Oga et al., 2011, and Ross et al., 2014).
- A literature review capturing data from 1,608 patients revealed that 49% of ACC tumors are positive for p53 expression and that p53-positivity correlates with solid histology, local recurrence, distant metastasis, nerve infiltration and poorer 5-year overall survival (Li et al., 2017).
Preclinical Evidence
- Treatment of low passage ACC cells with the MDM2 inhibitor MI-773 stabilizes levels of p53, phosphorylated p53 Serine 392 and the p53-target gene p21. MI-773 treatment also promotes apoptosis and tumor regression in ACC PDX models (Warner et al., 2016).
- MI-773 treatment sensitizes ACC cells to cisplatin treatment through a mechanism involving p53-mediated upregulation of pro-apoptotic genes, such as BAX and PUMA, and depletion of cancer stem cells (as defined by high levels of ALDH and CD44). Neoadjuvant MI-773 treatment followed by surgical resection of ACC PDX tumors elicited a durable anti-tumor response for up to 300 days (half the standard lifetime of a mouse; Nor et al., 2017).
- MDM2 inhibition via AMG 232 radiosensitizes a p53-WT ACC PDX model (Prabakaran et al., 2017).
Clinical Evidence
- A phase I clinical trial testing the MDM2 inhibitor APG-115 showed stable disease in 2 ACC patients (Zhang et al., ASCO 2019).
- A phase I/II trial (NCT03781986) testing MDM2i APG-115 in ACC patients is currently recruiting; preliminary results show a 17% objective response rate and median progression-free survival of 11.4 months with 91% of patients progression-free at >6 months (Pearson et al., EORTC-NCI-AACR 2022).
AXL is a cell surface receptor tyrosine kinase that regulates cell survival, cell proliferation, migration, differentiation and anti-inflammation. Downstream of the activated receptor lies the PI3K, GRB2 and Toll-like receptor signaling pathways, among others.
Molecular evidence
RNA/protein
- AXL activity is elevated in ACC cells and may modulate FGFR1-driven AKT signaling (Humtsoe et al., 2021).
- AXL levels (RNA transcript and protein) are increased in the ACC-II molecular subtype, also characterized by upregulation of EGFR, HER2, MET, and PI3K/AKT and RAS signaling pathways (Ferrarotto et al., 2021).
- Sixty percent of ACC tumors express high levels of AXL (defined as >25% positive cells) (Humtsoe et al., AACR 2022).
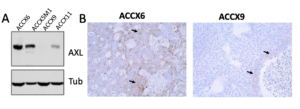
Preclinical evidence
- AXL inhibition (via siRNA or antibody drug conjugate; ADCT-601) induces a potent dose-dependent cytotoxic effect in ACC cells (Humtsoe et al., AACR 2022).
- In an AXL-high PDX model, a single dose of AXL-targeting ADC (ADCT-601) induced a durable, partial response (>50% tumor growth inhibition) in 4/5 (0.5 mg/kg dose level) and 5/5 (1 mg/kg dose level; 1 animal with complete response). More modest anti-tumor activity was observed with ADCT-601 in a PDX model expressing lower levels of AXL (Humtsoe et al., AACR 2022).
Clinical Evidence
- A 2024 case report describes the first ACC patient treated with mecbotamab vedotin (BA3011, an AXL-targeting, conditionally active biologic antibody-drug conjugate). After confirming a high level of AXL expression in the patient’s tumor, the patient received BA3011 treatment, which elicited a 25% reduction in target lesion growth (Hoff et al., 2024).
- A phase I/II trial (NCT03425279) testing a conditionally active biologic (CAB) AXL-targeted antibody-drug conjugate (CAB-AXL-ADC) is actively recruiting ACC patients (biopsy required to confirm AXL expression).
B7-H4 is a coinhibitory molecule that negatively regulates T cell immune responses and may contribute to tumorigenesis through multiple mechanisms. B7-H4 has been shown to be abnormally expressed in a variety of cancers.
MOLECULAR EVIDENCE
- Using Reverse-Phase Protein Array (RPPA), B7-H4 was found to be overexpressed in ACC-I tumors, alongside MYC, cell cycle regulators (PAR, CHK2, CHK1, CDK7), NOTCH1 and apoptosis markers (BCL2, BIM) (Ferrarotto et al., 2021).
- Overexpression of B7-H4 protein in ACC-I tumors was confirmed using imaging mass cytometry (IMC) and standard immunohistochemistry (IHC) analyses. B7-H4 expression was localized to the core of ACC-I tumors and associated with T-cell restriction to the stroma (Guimaraes de Sousa et al., 2023).
Preclinical Evidence
- A single dose of AZD8205 (B7-H4-directed antibody-drug conjugate that uses a topoisomerase 1 inhibitor payload) inhibited tumor growth and led to tumor regressions in 100% of ACC-I-type PDXs tested. Complete responses were also observed in some AZD8205-treated animals with ACC-I tumors. No anti-tumor activity was observed in an ACC-II-type PDX treated with AZD8205 (Guimaraes de Sousa et al., 2023).
clinical Evidence
- High IHC staining for B7-H4 was associated with poor prognosis (p=0.01; HR=2.54), even after controlling for stage and histology (Guimaraes de Sousa et al., 2023).

- A phase I trial (NCT05377996) testing a B7-H4 ADC (XMT-1660) is currently open to ACC patients.
TROP-2 is a cell surface protein involved in calcium signaling. High levels of TROP-2 have been found in a variety of cancers.
Molecular evidence
RNA/protein
- Positive TROP-2 staining was observed in 95.5% (21/22; low=13.6%; moderate=54.5%; high=27.4%) of ACC cases using immunohistochemical staining and MALDI-mass spectrometry (Wolber et al., 2021). Also see Linxweiler et al., 2024, for examples of elevated TROP2 staining in ACC samples.
- TROP2 IHC performed across a cohort of 165 ACC tumors revealed the highest TROP2 levels in tumors lacking solid histology (typical of ACC-II, confirmed by RNAseq). Interestingly, in cribriform and tubular samples, TROP2 staining was highest in ductal/epithelial cells but was lost in (ductal/epithelial-dominant) solid tumors, potentially indicating that paracrine signaling maintains TROP2 expression in tumors with dual cell populations (Siqueira et al., 2025).
- TROP2 IHC was conducted across the 2022 ACC PDX tissue microarray using the TROP2 antibody from Cell Marque (clone EP431; catalog #465R). A PDF of the TROP2 stains is also available: TROP2 IHC PDX.
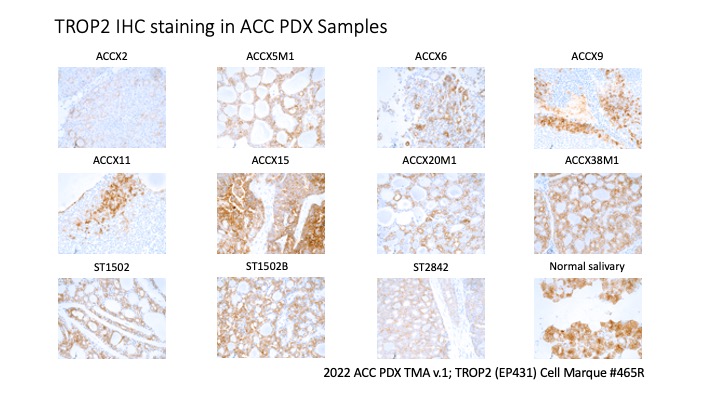
Preclinical Evidence
- The viability of a TROP2-positive PDX-derived ACC cell line was negatively impacted upon treatment with sacituzumab govitecan (antibody-drug conjugate targeting TROP2 (Siqueira et al., 2023).
clinical Evidence
- A phase 1 trial (NCT05884320) testing sacituzumab govitecan (antibody-drug conjugate targeting TROP2) is currently open to recurrent/metastatic ACC patients.
- A phase 1 trial (NCT05941507) testing LCB84 (antibody-drug conjugate targeting TROP2) and anti-PD-1 monoclonal antibody is currently open to recurrent/metastatic ACC patients. Pre- and on-treatment biopsies are required if deemed medically feasible and safe).
Selected publications describing cell surface protein expression in ACC are listed below. Additional efforts to characterize the ACC cell surfacesome are currently underway.
Nectin-4 (Nectin cell adhesion protein 4)
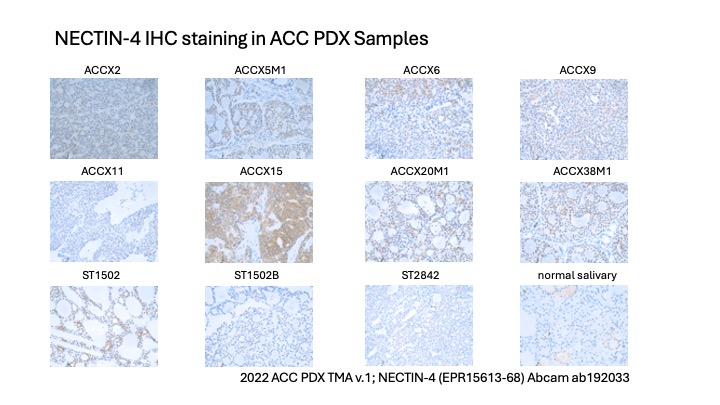
- Nectin-4 is a cell surface protein involved in calcium-independent cellular adhesion. A Nectin-4-directed antibody drug conjugate Enfortumab vedotin-ejfv was approved for locally advanced or metastatic urothelial cancer in 2019. Moderate or high Nectin-4 expression was observed in 30.7% of 26 ACC tumors analyzed (Mayer et al., 2023)
- Nectin-4 IHC was conducted across the 2022 ACC PDX tissue microarray using the Nectin-4 antibody from Abcam (clone EPR15613-68; catalog #ab192033). A PDF of the Nectin-4 stains is also available: Nectin4 IHC PDF.
- A Phase II trial testing enfortumab vedotin (ADC targeting Nectin-4) is currently open to recurrent/metastatic ACC patients (NCT06891560).
Fra (Folate Receptor alpha)
- Folate receptor alpha binds folate, which is required for various metabolic processes and has been found upregulated in multiple cancers. In one study, FRa expression was detected in 47% (low=21%; moderate=19%; high=2%) of ACC cases (n=43), with staining localized at the cell membrane and throughout the cytoplasm. Weak FRa staining was detected in normal surrounding tissue in 22% of cases; however, uptake of a PET tracer targeting folate receptor has been reported to be low in salivary glands (Schnoell et al., 2022 and Gnesin et al., 2020).
Mucins
- The mucin family of proteins produce gel-like secretions essential for cell lubrication and barrier protection and are frequently found overexpressed in cancers. In limited analyses, 73% (8/20) of adenoid cystic cancers stained positive for MUC3, but were negative for other mucin family members (MUC1, MUC3, MUC5AC, MUC6 and MUC16; Lee et al., 2005 and Wolber et al., 2022).
Fibroblast Activation Protein (FAP)
- FAP is a type II membrane-bound glycoprotein that is frequently overexpressed in cancer-associated fibroblasts. FAP inhibitor-based radiopharmaceuticals are currently in development for PET imaging and radiotherapy (2022 review). 68 Ga-FAPI PET/CT in 2 ACC patients revealed precise detection of locoregional recurrence in one case and intense FAP uptake of widespread metastases in the second case (Civan et al., 2023).
Claudins
- Caludins are a tetraspan membrane protein involved in constructing tight junctions between cells and maintaining cell polarity. Some types of claudins are dysregulated in cancer. Analysis of 12 ACC lung tumors for IHC of various claudin proteins revealed high levels of CLDN2 and CLD4 expression (although the age of the FFPE sample somewhat influenced these results). In contrast, ACC tumors were absent for CLDN1 expression (Gyulai et a., 2023).
BCL-2 is a central member of the BCL-2 family (including BAX and BAK) of pro-apoptotic proteins that work at the mitochondrial membrane to inhibit initial steps of cell death (membrane permeabilization and cytochrome C/ROS release). Overexpression of BCL-2 has been implicated in cancer (for ex. Lymphoma) and BCL-2 inhibitors are used clinically to treat chronic lymphocytic leukemia and small lymphocytic lymphoma.
Molecular evidence
RNA/protein
- BCL2 levels (RNA and protein) are increased in ACC-I subgroup alongside activating NOTCH mutations and high MYC expression (Ferrarotto et al., 2021).
Preclinical evidence
- BCL2 inhibitor ABT-199 treatment alone or in combination with NOTCH inhibitor AL101 exerts a strong anti-tumor effect in NOTCH-mutant ACC PDX models (Ferrarotto et al., ESMO 2021).
clinical evidence
- High BCL-2 expression was significantly associated with worse OS, even after adjusting for histology, stage, and other grouped variables (Sousa et al., 2023).
PSMA (Prostate-Specific Membrane Antigen) is a transmembrane glycoprotein that is highly expressed on prostate cancer cells, but can also be detected at high levels in other cancers and at lower levels in certain normal tissues, including the salivary glands.
Historically, PSMA levels/expression have been used as a biomarker to predict prostate cancer aggressiveness and, more recently, as a therapeutic targeting agent using small-molecule radiolabeled ligands that can bind the cytoplasmic domain of PSMA and be internalized via clathrin-coated pits (Donin et al., 2018).>).
Molecular evidence
RNA/protein
- PSMA expression, as assessed by PSMA-radiolabeled ligand uptake and immunohistochemistry, is observed in the majority of primary, locally recurrent and distant metastatic ACC tumors (Lutje et al., 2016, Klein Nulent et al., 2017, Konig et al., 2017, de Keizer et al., 2017, van Boxtel et al., 2020, Klein Nulent et al., 2020 and Shamim et al., 2023)
- PSMA immunohistochemistry is not predictive of ligand uptake (Klein Nulent et al., 2017, Konig et al., 2017 and van Boxtel et al., 2020).
Clinical evidence
- In a phase II imaging study, the maximum standardized uptake value of a PSMA radiolabeled tracer ranged from 1.1 to 30.2 with a tumor/liver ratio>1 in 13/14 (93%) ACC patients (van Boxtel et al., 2020).
- PSMA uptake can vary at both the intra-patient and inter-metastatic levels (van Boxtel et al., 2020).
- 177Lu-PSMA treatment in an ACC patient with widespread bone metastases led to a significant reduction in pain; however, the patient died before follow-up tumor scans could be captured (Simsek et al., 2019 and reviewed in Uijen et al., 2021).
- A phase II trial testing 177Lu-PSMA-I&T (NCT04291300) in 10 ACC patients showed stable disease as the best response, with a median progression-free survival of 6.7 months and progression-free duration reaching 30% of patients for >3 months (van Herpen et al., 2023 ASCO abstract).
- A phase II therapeutic trial of 177Lu-EB-PSMA-617 is enrolling ACC patients in China (NCT04801264).
- A phase I therapeutic trial testing P-PSMA-101 CAR-T therapy (NCT04249947) enrolled several ACC patients; awaiting results.
Chromatin modifying genes include classes of proteins that alter the chemistry of histones, thereby influencing gene expression by regulating access of condensed DNA to transcription machinery. These protein classes are histone acetyltransferases (HATs), deacetylases, methyltransferases, kinases, and ATP-dependent protein complexes that move, remove or restructure nucleosomes.
Deregulated activity of chromatin modifying genes has been implicated in cancer through a variety of mechanisms, including disrupted expression of oncogenes and tumor suppressor genes.
Molecular Evidence
DNA
- Many chromatin remodeling genes have been reported to be mutated at low frequency. Genes mutated in >5% of patients are highlighted in red (Ho et al., 2013 and Ho et al., 2019).
- SWI/SNF: SMARCA2 (<1-8%), ARID1A (3-15%), ARID1B (10%), SMARCE1 (<1-2%), ATRX (2-3%), SRCAP (<1-2%)
- Histone acetyltransferases: CREBBP (7-12%), EP300 (2-10%), KAT6A (2-4%), BRD1 (<1-2%), MORF4L1 (<1-2%), KANSL1 (<1-2%)
- Histone methyltransferases: KMT2C/MLL3 (2-12%), KMT2D/MLL2 (15%), CMTR2/FTSJD1 (<1-5%), SETD2 (3-5%), NSD1 (2-3%)
- Histone demethylases: KDM6A (7-15%), KDM6B (<1-2%), ARID5B (<1-2%), JMJD1C (<1-2%)
- Histone deacetylate subunits: BCOR (2-12%), BCORL1 (2-3%), ARID4B (<1-3%)
- Some chromatin remodeling gene mutations occur at residues in protein domains required for their catalytic activity. For example, SMARCA2 mutations cluster within the helicase C domain and CREBBP mutations cluster in the KAT11 histone acetylation domatin (Ho et al., 2013).
- Mutations in some chromatin modifying genes are enriched in recurrent/metastatic compared to primary ACC tumors, including KDM6A (15.2% vs. 3.4%), KMT2C/MLL3 (14.3% vs. 4%) and ARID1B (14.1% vs. 4%). These gene mutations tend to be inactivating and occur in tumor hotspot regions, suggesting they may play a role in driving cancer progression (Ho et al., 2019).
- A summary of epigenetic alterations observed in ACC tumors can be found in Manou et al., 2023.
Preclinical Evidence
- HDAC inhibitor (trichostatin A and vorinostat) treatment reduces MYB expression in some cancer cell contexts (Chambers et al., 2003 and Claerhout et al., 2011).
- Treatment of ACC primary cells with the HDAC inhibitor vorinostat and cisplatin reduces viability by promoting senescence (Almeida et al., 2017).
- Entinostat treatment inhibits the growth of ACC PDX tumors alone or in combination with a low dose of cisplatin (Mangold et al., 2018).
- Overexpression of KDM6A mutants observed in ACC tumors increases cell proliferation and abrogates KDM6A’s H3K27me3 demethylase activity (Ho et al., 2013.)
Clinical Evidence
- Phase 1
- A clinical trial testing vorinostat in patients with advanced solid tumors revealed 1 ACC patient with a partial response and 4 patients with stable disease (Ramalingham et al., 2010).
- A clinical trial testing chidamide in patients with advanced solid tumors and lymphomas showed 1 ACC patient (3.2%) with a partial response (Dong et al., 2012).
- Phase 2
- A clinical trial testing vorinostat in ACC patients with locally advanced, recurrent or metastatic ACC patients showed a partial response in 2 patients (6.7%) with a response duration of 53 and 7.2 months, 1 patient (3.3%) with a minor response and stable disease in 27 patients (90%; Goncalves et al., 2017).
- Of the 12 recurrent/metastatic ACC patients included in a phase 2 clinical trial testing vorinostat plus pembrolizumab, 1 patient (8.3%) experienced a partial response. Of the 25 salivary gland patients, 14 (56%) experienced stable disease and the median progression free survival was 6.9 months (Rodriguez et al., 2019)
Immune checkpoints are inhibitory regulators of the immune system that are crucial to maintaining self- tolerance, preventing autoimmunity, and controlling the duration and extent of immune responses in order to minimize collateral tissue damage. These immune checkpoints are often overexpressed on tumor cells or on non-transformed cells within the tumor microenvironment, and compromise the ability of the immune system to mount an effective anti-tumor response.
PD-1 is a member of the CD28/CTLA-4 family of cell surface receptors that is mainly expressed on immune-related T cells, B cells and macrophages. Binding of PD-1 to its two ligands, PD-L1 and PD-L2, activates an immune checkpoint response that inhibits the immune system by suppressing T cell function. PD-1-mediated immune checkpoints are thought to be important in preventing autoimmunity.
Many cancers upregulate the expression of PD-1 ligands (most commonly PD-L1, but also PD-L2) in order to inhibit immune checkpoint control and escape anti-tumorigenic immune surveillance. Clinical development of immune checkpoint blockade antibodies targeting PD-1 inhibit the interaction between PD-1 and PD-L1/2 and promote anti-tumor activity by reactivating T cell-driven immune responses. In some cancers, it is not clear which specific molecular biomarkers (PD-L1/2 expression, mutagenic load or lineage) best predict clinical response to PD-1 inhibition.
Molecular Evidence
DNA/RNA/protein/cellular
- Immune profiling of ACC tumors suggests they are immunologically “cold”, showing little to no PD-L1 expression (although approximately 60-75% of ACC tumors express some PD-L2), few immune cell infiltrates, low tumor mutational burden and presence of M2-polarized macrophages and myeloid-derived suppressor cells. Loss of heterozygosity at the HLA locus, which may also lead to immune evasion, has also been observed in ACC tumors (Sridharan et al., 2016, Wolkow et al., 2020 and Linxweiler et al., 2020).
- Expression of T cell checkpoint proteins: PD1, CTLA4, LAG3, TIM3 and TIGIT is low in ACC tumors (Mosconi et al., 2019, Linxweiler et al., 2020). ACC tumors that do express LAG3 are associated with shorter event free survival (Arolt et al., 2020).
- T cell infiltration is lower in recurrent/metastatic ACCs compared with non-recurrent/metastatic tumors (Mosconi et al., 2019 and Linxweiler et al., 2020).
- SNV-neoantigen and gene fusion-neoantigen count correlate with immune cell infiltration (Linxweiler et al., 2020).
- Together, these findings suggest that treatment with any single checkpoint inhibitor is unlikely to be successful in ACC patients and has spurred a series of clinical trials testing checkpoint inhibitors in combination with radiation, chemotherapy or other targeted/immunotherapies.
Preclinical Evidence
- MYB-NFIB peptides chosen based on ACC tumor-derived RNA sequencing and predicted HLA binding binding affinity are able to stimulate IFN-Ɣ secretion when expressed in the patient’s T cells or healthy donor T cells (Yang et al., 2019).
Clinical Evidence
- Chemoradiation has been shown to increase intratumoral recruitment of CD8+ effector T cells, decrease regulatory T cell infiltration and promote a systemic humoral response in ACC patients (Sridharan et al., 2016,). However, preliminary results from a phase II clinical trial combining Pembrolizumab with or without radiation in recurrent/metastatic ACC patients did not yield objective responses and no difference in stable disease or progression-free survival was seen between treatment arms (Schoenfeld, ASCO 2019).
- Clinical trials testing PD-1 inhibitors Pembrolizumab and Nivolumab alone or in combination with Ipilumumab (CTLA-4) or Vorinostat have not shown robust anti-tumor activity in ACC with an objective response rate of 0-9% and progression-free survival ranging 4-6 months (Fayette et al., ASCO 2019, Tchekmedyian et al., ASCO 2019, PD-1 alone ASCO 2019 and Rodriguez et al., 2020).
- Phase II trials testing TKIs with immune checkpoint inhibitors have been tested in ACC patients.
- Axitinib (TKI) + Avelumab (PD-L1i) was tested in 28 ACC patients and showed an 18% response rate, 7.3 months progression-free survival, with 57% of patients displaying stable disease for >6 months (Ferrarotto et al., 2023).
- Lenvatinib (TKI) + Pembrolizumab (PD-1i) was tested in 17 ACC patients and showed a 6% response rate, with 65% of patients displaying stable disease for > 6 months (Mohamadpour et al., 2023 ASCO abstract).
- Exceptional responses to immunotherapy have been reported in a very limited number of ACC patients. These patients are being studied to better understand why their tumor or tumor microenvironment makes them exquisitely sensitive to immunotherapy.
PRMT5 is an enzyme that catalyzes symmetric dimethylation of arginine residues on histones H3 and H4, marks typically associated with transcriptional repression. The molecular mechanisms that underlie ACC cell sensitivity to PRTM5 inhibition are not yet understood.
Preclinical evidence
- PRMT5 inhibitor PRT543 showed anti-tumor activity in ACC PDX models (Bhagwat et al., 2020 and McKean et al., 2021) and inhibited MYB and NOTCH signaling (Carter et al., 2021).
clinical evidence
- Three out of fourteen (21.4%) ACC patients experienced a partial response while being treated with GSK3326595 (PRMT5 inhibitor) during a phase 1 clinical trial (Siu et al., 2019).
- Five out of seven (71.4%) ACC patients experienced stable disease for some duration of time while being treated with PRT543 in a phase 1 clinical trial (McKean et al., 2021).
- Janssen’s PRMT5 inhibitor JNJ64619178 (Onametostat) showed a 9% partial response rate in 11 ACC patients (Villar et al., ESMO 2020).
IGF-1R (Insulin-like Growth Factor Receptor 1) is a transmembrane tyrosine kinase receptor that undergoes autophosphorylation and activation upon binding to its ligands insulin-like growth factor 1 or 2 (IGF-1, -2). IGF-1R signaling is involved in regulating normal organ growth and development, and downstream substrates include the RAS/MAPK and PI3K/AKT pathways.
IGF-1R expression is upregulated in a number of cancers and elevated IGF-1 serum levels has been linked to increased epithelial cancer risk. Forced IGF-1R expression can promote tumor growth in vivo and inhibition of IGF-1R has been shown to suppress mitogenic activity and anchorage-independent growth of cells. While the specific mechanisms by which IGF-1R promotes tumorigenesis remain unclear, the kinase domain is thought to be required for transformation. (Yakar et al., 2005).
Molecular Evidence
DNA
- Mutations in IGF-BP2 (Ho et al., 2013) and amplification of IGF-1R (Ross et al., 2014) have been observed in ACC tumors.
- Activating IGF1R hotspot non-frameshift insertion mutations occurring in the hinge region of the fibronectin type 3 domain and tyrosine kinase domain were observed in 3.5% (55/1564) of ACC samples (Margolis et al., 2022).
RNA/protein
- IGF-1R is frequently upregulated and phosphorylated in ACC tumors (Dr. Chris Moskaluk, UVA, personal communication).
- Cytoplasmic and nuclear levels of IGF-1Rb are higher in ACC samples compared to adjacent salivary glands (120 samples analyzed; Ouyang et al., 2017).
Preclinical Evidence
- Expression of IGF-1R and IGF-1, -2 is regulated by MYB in a variety of cancer cells (Kim et al., 2009, Tanno et al., 2002 and Reiss et al., 1991).
- MYB expression predicts figitumumab (IGF1R antibody) sensitivity across a panel of 93 cancer cell lines (Pavlicek et al., 2013).
- MYB-NFIB expression is regulated by IGF-1R in ACC cell cultures. Combined inhibition of IGF-1R (via linsitinib), EGFR (via gefitinib) and MET (via crizotinib) synergistically inhibits growth in ACC cells both in vitro and in vivo (Andersson et al., 2017).
- ACC PDX models, particularly those with reduced IGF-1R gene signature at baseline, are more sensitive to figitumumab treatment (Calvo et al., 2017).
Clinical Evidence
- One ACC patient, predicted to be sensitive to figitumumab in a personalized ACC tumorgraft, experienced minor yet durable (6 months) tumor shrinkage while being treated with figitumumab (Morelli et al, 2012).
- One ACC patient was treated for over 1 year during a phase Ib trial for figitumumab combined with sorafenib (Mahadevan et al., 2014).
- In a phase I clinical trial testing figitumumab (IGF-1R antibody) and dacomitinib (EGFR inhibitor), 1 ACC patient achieved a partial response and continued on treatment for 1.5 years. Stable disease was observed in 2 additional ACC patients (Calvo et al., 2017).
NFIB (Nuclear Factor IB) belongs to the nuclear factor I (NFI) family of transcription factors that also includes NFIA, NFIC and NFIX. NFI proteins are regulated in a tissue-specific manner and bind DNA as hetero- or homodimers to activate or repress transcription, depending on the cellular context. NFIB knockout mice are embryonic lethal due to lung and brain developmental defects (Steele-Perkins et al., 2005), and NFIB-mediated transcription has been linked to regulation of stem cell behavior in hair follicles (Chang et al., 2013), embryonic development of submandibular glands (Mellas et al., 2015) and mammary gland development (Murtagh et al., 2003).
NFIB posses tumor suppressive functions in a wide range of cancers, including benign prostatic hyperplasia (Grabowska et al., 2016), metastatic osteosarcoma (Mirabello et al., 2015), glioblastoma (Stringer et al., 2016) and cutaneous squamous cell carcinoma (Zhou et al., 2015). NFIB loss is linked to enhanced stem cell proliferation (Chung et al., 2013), increased CDK4/6 expression (Zhou et al., 2015) and upregulation of EZH2 (Piper et al, 2014), the last of which is a predictor of poor clinical outcome in ACC patients (Vekony et al., 2008). NFIB overexpression has also been shown to limit the transformation of normal fibroblasts by several nuclear oncogenes, including MYC (Schuur et al., 1995), through unknown mechanism. Interestingly, NFIB amplification also can promote small cell lung cancer initiation (Dooley et al., 2011) and progression by altering chromatin accessibility (Semenova et al, 2016, Denny et al, 2016). Together, these findings reinforce that NFIB may have opposing functions in different cancers.
Recurrent fusions of NFIB have been found with HMGIC in pleomorphic adenomas of the salivary gland (Geurts et al., 1998) and with HMGA2 in lipomas (Nilsson et al., 2005).
Molecular Evidence
- Chromosomal rearrangements between MYB/L1 and the 3’ end of the NFIB gene are frequently detected in ACC (Persson et al., 2009 and Mitani et al., 2016).
- The 5’ end of the NFIB has been found fused to other genes, including XRCC4, NKAIN2, PTPRD and AIG1 (Mitani et al., 2016). The significance of these gene fusions in ACC pathogenesis is unclear.
- A hallmark of all MYB/L1-NFIB translocations is that they bring super-enhancers from the NFIB locus proximal to the MYB or MYBL1 genes (Drier et al., 2016).
- Inactivating mutations in the non-rearranged NFIB allele can co-occur with MYB-NFIB fusions in ACC (Ho et al., 2013). It remains unclear whether loss of one or both NFIB alleles contributes to ACC pathogenesis.
RNA/protein
- The NFIB transcription factor is highly expressed in normal salivary tissue (Drier et al., 2016).
- ACC-derived MYB/L1-NFIB translocations often generate a fusion transcript that includes a small portion of NFIB’s C-terminus (Mitani et al., 2011, Togashi et al., 2018). It is not clear whether the NFIB residues that are included in the transcript (in some cases only the last 5 amino acids) contribute to altered MYB function.
- NFIB is overexpressed in a subset of ACC tumors (irrespective of NFIB rearrangement status) compared to normal salivary tissue (Rettig et al., 2016).
CDK9 (Cyclin Dependent Kinase 9) is a member of the P-TEFb (positive transcription elongation factor) complex that regulates RNA polymerase II activity and the transcriptional elongation of genes. CDK9 binds to cyclin T and phosphorylates the C-terminal domain of RNA Pol II.
Given CDK9’s role as a master regulator of transcription, cancers driven by aberrant activity of oncogenic transcription factors like MYC and MYB are hypothesized to be particularly vulnerable to CDK9 inhibition.
Preclinical Evidence
- Treatment of MYB-overexpressing breast cancer cells with CDK9 inhibitors, such as flavopiridol/alvocidib, inhibits MYB expression due to prolonged transcriptional pausing at intron 1 of the MYB locus (Mitra et al., 2012 and Mitra et al., 2016).
- CDK9 inhibitor PRT2527 showed anti-tumor activity in an ACC PDX model (Zhang et al., 2021).
- CDK9 inhibitor KB-0742 showed anti-tumor activity in multiple ACC PDX models (McKeown et al., 2024).
Clinical Evidence
- One ACC patient achieved stable disease ≥6 cycles (3-week intervals) upon treatment with the CDK9 inhibitor, CYC065 (Khanh et al., 2018).
The bromodomain and extraterminal domain (BET) family of proteins (BRD2, BRD3, BRD4 and BRDT) harbor a conserved bromodomain involved in protein interactions and binding to acetylated lysines, extra terminal domain and divergent C-terminal recruitment domain. BET proteins act as “chromatin readers” of lysine acetylation and regulate gene expression by recruiting factors to chromatin, such as PTEFb and other transcription-modifying proteins (Fu et al., 2015).
Deregulated BET activity can lead to inappropriate acetylation and transcriptional activation of oncogenes. Translocations involving BRD4 have been detected in NUT midline carcinoma and BET family members have been shown to deregulate expression of cancer genes in leukemia (Muller et al., 2011). Mechanistically, binding of BRD4 and the Mediator transcriptional complex to large regions of chromatin have been suggested to form super enhancer regions that promote the expression of oncogenic drivers such as MYC (Loven et al., 2013).
Molecular Evidence
RNA/protein
- In ACC tumors, BRD4 binds at super enhancers that are repositioned following chromosomal translocation of NFIB, TGFBR3 or RAD51B near the MYB locus (Drier et al., 2016).
Preclinical Evidence
- BET inhibitors suppress MYB expression in acute myeloid leukemia cells (Roe et al., 2015).
- The BET bromodomain inhibitor JQ1 inhibits tumor growth and expression of MYB and MYB target genes in low-grade ACC PDXs (Drier et al., 2016).
The PI3K (Phosphoinositide 3-Kinase) pathway is driven by three kinases (PIK3CA, PIK3CB, PIK3CD), which are activated by cell surface receptor tyrosine kinases and regulate cell growth and survival pathways through their downstream substrates, which include AKT and mTOR. The PI3K pathway is negatively regulated by the PTEN phosphatase (Vanhaesebroeck et al., 2012).
Aberrant activation of the PI3K pathway (either via mutation/amplification of PIK3CA/B/D, AKT or mTOR or deletion of PTEN) has been implicated in a wide range of cancers and has led to the development of inhibitors targeting different nodes within the pathway (Yang et al., 2019).
Molecular Evidence
DNA
- A subset of ACC tumors harbor alterations in PI3K pathway genes, including PIK3CA (6%), PTEN (3%), AKT1 (2%) and MTOR (2.6%). Some of these mutations/amplification/deletions occur in tumor hotspot regions and may predict sensitivity to PI3K/AKT/MTOR inhibitors (Ettl et al., 2012, Ho et al., 2013, Stephens et al., 2013 and Ho et al., 2019).
RNA and protein
- Increased levels of phosphorylated AKT (PI3K target) are associated with a higher relapse rate in ACC patients (Völker et al., 2009).
- Hemizygous loss of PTEN or reduced PTEN expression is observed in 10-15% of ACC tumors (Ettl et al., 2012).
- Increased expression of PI3K pathway genes correlate with low immune cell infiltration in ACC tumors (Sridharan et al., 2016).
Preclinical Evidence
- Inducible overexpression of AKT3 in the salivary glands (via MMTV) drives tumor formation in all major salivary glands with 100% penetrance. Tumors that form in this model show histology consistent with ACC and stain positive for smooth muscle actin (Zboray et al., 2018).
- Co-deletion of PTEN and SMAD4 in murine salivary glands leads to the development of salivary gland malignancies, including ACC- as evidenced by hallmark histology and positive staining for keratin 5, smooth muscle actin and p63. ACC tumor formation occurred in 6/11 animals in the cohort. Other histologies include salivary ductal (3/11) and salivary adenocarcinoma (2/11; Cao et al., 2018). Of note, concurrent loss of PTEN and SMAD4 has not been observed in a significant proportion of ACC tumors (Ho et al., 2019).
- Several PI3K/AKT/mTOR inhibitors have shown significant anti-tumor activity in ACC PDX models (ACCRF-generated data, unpublished).
Clinical Evidence
- A phase II clinical trial testing mTOR inhibitor everolimus in recurrent/metastatic ACC patients showed no objective responses, but stable disease with some tumor shrinkage in 15 of 34 (44%) patients and stable disease lasting greater than 6 months in 13 patients (38%). Median progression free survival was 11.2 months (Kim et al., 2014).
- A phase II clinical trial of the AKT inhibitor MK-2206 showed stable disease as the best response in 13/14 recurrent/metastatic ACC patients and a median progression free survival of 9.2 months. In serial biopsies, MK-2206 treatment led to a reduction in AKT signaling and MYB expression in 4 of 5 cases tested (Ho et al., ASCO 2015).
- Testing of the HIV protease inhibitor drug nelfinavir, which has also been shown to inhibit AKT signaling, in a phase II clinical trial in recurrent ACC patients led to a median progression free survival of 5.5 months but no objective responses (Hoover et al., 2015).
The WNT/beta-catenin pathway is activated in response to WNT ligand binding to cell surface receptors FRIZZLED or LRP5/6, which triggers a series of events that inhibit the GSK-3 beta/CK1/APC/Axin destruction complex and allows stabilization and nuclear translocation of beta-catenin. Within the nucleus, active beta-catenin regulates transcription by binding to LEF/TCF transcription factors, displacing co-repressors and recruiting additional transcriptional co-activator proteins. WNT/beta-catenin signaling has been implicated in stem cell regulation, bone development (MacDonald et al., 2009) and salivary gland stem/progenitor cell activity during postnatal development and regeneration (Hai et al., 2010).
Mutations in beta-catenin or deletion/loss-of-function mutations in APC or Axin are frequently detected in cancer especially in the colon, breast, pancreas and head and neck cancers (Clever and Nusse, 2012). Within head and neck cancers, WNT/beta-catenin-mediated transcription has been implicated in cancer stem cell function, metastasis and chemoresistance (Aminuddin and Ng, 2016).
Molecular Evidence
DNA
- CTNNB and APC genes are altered in approximately 2.5% and 5% of ACC tumors, respectively. A subset of APC alterations include putative driver truncating mutations (Ho et al., 2019).
RNA/protein
- Wnt/beta-catenin genes casein kinase 1, epsilon and frizzled-7 are upregulated in ACC tumors (Frierson et al., 2002).
- Wnt/beta-catenin signaling has also been proposed to regulate MYB in some contexts (Ramsay and Gonda, 2008).
- Increased expression of Wnt/beta-catenin pathway genes correlate with low immune cell infiltration in ACC tumors (Sridharan et al., 2016).
The EGFR (Epidermal Growth Factor Receptor) family of cell surface receptors consists of four tyrosine kinases, EGFR (ERB-1), HER2/c-neu (ERB-2), HER3 (ERB-3) and HER4 (ERB-4). Upon binding to their ligands (EGF, transforming growth factor a, etc.) EGFR transitions from an inactive monomer to an active homodimer, which stimulates its intracellular tyrosine kinase activity. Downstream substrates of EGFR include the PI3K, STAT and RAS pathways, and EGFR signaling has been implicated in mammary gland development.
Activating mutations and amplifications in the EGFR gene have been detected in a wide range of solid tumors, including lung, breast, and head and neck cancers.
Molecular Evidence
DNA
- A small percentage (2%) of ACC patients have alterations in the EGFR gene (Saida et al., 2018; Ho et al., 2019).
RNA/protein
- EGFR expression is frequently upregulated in ACC tumors (Vered et al., 2002; Macarenco et al., 2008; Locati et al., 2009; Schneider et al., 2016).
- The average EGFR positive score in a cohort of ACC samples was 51.6%, and the average cumulative EGFR score in ACC was 80 (Fujiwara et al., 2024).
- Three out of 15 ACC tumor samples displayed overexpression of EGFR using immunohistochemistry (Linxweiler et al., 2024).
Preclinical Evidence
- Combined inhibition of IGFR, EGFR, and MET reduces MYB expression, drives ACC cell differentiation, and synergistically inhibits ACC PDX tumor growth (Andersson et al., 2017).
- Dual targeting of EGF and VEGFR kinases via AEE788 inhibits ACC cell growth in vitro and in vivo (Younes et al., 2008).
Clinical Evidence
- Three phase II clinical trials testing different EGFR inhibitors: Gefinitib (Jakob et al., 2015), Cetuximab Locati et al., 2009 and Lapatinib (Agulnik et al., 2007) in advanced salivary cancer patients (including ACC) showed a median progression-free survival of 4.3 months, 6 months, or 3.5 months, respectively, and no objective responses. No statistically significant correlation between EGFR expression and clinical response was observed in the aforementioned studies; however, several patients with high levels of EGFR did show significant progression-free survival.
- In a Phase I study of Figitumumab combined with Dacomitinib, one ACC patient achieved a partial response and continued on treatment for 1.5 years (Calvo et al., 2016).
-
One metastatic ACC patient with histologically proven EGFR expression in their tumor showed stable disease for 17 months while receiving treatment with Gefitinib (Linxweiler et al., 2024).
The TRK pathway consists of TRKA, TRKB and TRKC transmembrane proteins, which are encoded by the NTRK1, NTRK2 and NTRK3 genes, respectively. TRK receptors are activated by binding to neurotrophin ligands and play a diverse set of roles in regulating the development and function of neuronal cells.
Oncogenic fusions involving NTRK genes have been observed in a variety of cancers, including colon, secretory breast, high-grade glioma and thyroid, and functionally lead to ligand-independent, constitutively active kinase activity (Amatu et al., 2019).
Molecular evidence
DNA
- Amplifications or missense mutations in NTRK1, -2 or -3 are observed in a small percentage of ACC tumors (1-6%; Ho et al., 2019). Fusions involving the NTRK genes have not been observed in ACC (Ivanov et al., 2012).
RNA/protein
- TRKC (phosphorylated and total protein) is highly expressed in primary ACC tumors and PDX samples. ACC samples also express NT-3, TRKC’s ligand (Ivanov et al., 2012).
- ACC samples co-express TRKC, p-ERK1/2 and BCL2 in their myoepithelial cell compartment (Ivanov et al., 2012).
Preclinical evidence
- ACC PDX models show varied sensitivity to TRK inhibition via AZD7451 (Ivanov et al., 2012).